
Die Diskussionen darüber, ob die neuartigen Corona-Impfstoffe wirklich sicher sind, häufen sich derzeit. Dabei müsste doch eigentlich alles ordnungsgemäß geprüft worden sein? Wie solche Prüfprozesse ablaufen, sollte man sich vielleicht einmal genauer ansehen.
Im Rahmen der Zulassung von Arzneimitteln gibt es verschiedene regulierende Institutionen. Diese werden jedoch zum Teil von der Industrie finanziert [1]. In den USA wird die Food and Drug Administration bereits seit 1992 von Pharmafirmen unterstützt, in 1993 waren es 29 Millionen US-Dollar, in 2016 884 Millionen [1]. In Europa wurde die European Medicines Agency (EMA) in 1995 zu 20 % durch die Pharmaindustrie finanziert, in 2010 dann schon zu 75 % und in 2022 zu 89 % [2]. Eine Studie des British Medical Journal hat den Einfluss der Pharmaindustrie auf die Regulierungsbehörden tabellarisch zusammengefasst (s. Abb. 1). Die Unternehmen, deren Produkte geprüft werden sollen, finanzieren demnach direkt und fast komplett die Institutionen, die dann später die Prüfung durchführen, und das nicht nur in Europa und den USA [3]. In Australien erreicht der prozentuale Anteil der Industrie am Budget der Therapeutic Goods Administration (TGA) sogar 96 %, und diese hatte in 2020 und 2021 im Schnitt neun von zehn Produkten zugelassen [3, 4]. Zudem kam es laut der Journalistin Rebekah Barnett bei der TGA zu Unregelmäßigkeiten bei den Meldungen von Todesfällen im Zusammenhang mit den neuartigen Corona-Impfstoffen [44]. Es ist laut Studien durch diese Verflechtungen ein Qualitätsabfall bei medizinischen Produkten zu verzeichnen [5, 6, 7, 8].
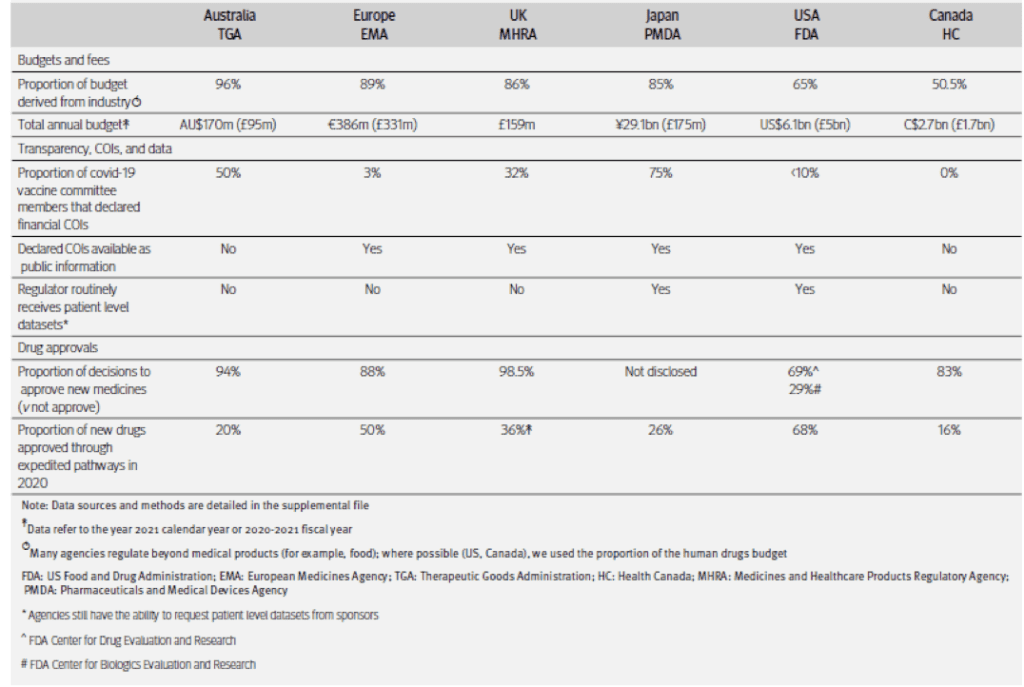
Abb. 1: Finanzierung der großen nationalen Regulierungsbehörden (Quelle: http://dx.doi.org/10.1136/bmj.o1538)
Auch die Beraterstäbe, welche den Regulierungsbehörden zur Seite stehen, sind gemäß dem Artikel teilweise finanziell mit den Pharmaunternehmen verflochten [5, 10], und Berater, die solche finanziellen Beziehungen zu Pharmaunternehmen pflegen, sprechen sich laut einer Studie häufiger für die Zulassung von Produkten aus [9]. Fünf von zehn Beratern, die für die TGA tätig waren, hatten gemäß einer journalistischen Anfrage einen Interessenkonflikt [3, 4], die Berater arbeiten häufig für die Firmen, die sie bewerten [3, 11, 12], und auch die Datenverarbeitung bei der Überprüfung der zuzulassenden Produkte wird von den industriellen Sponsoren erledigt [3, 13, 14, 15]. Im Rahmen der Zulassung der neuartigen Corona-Impfstoffe liegen der TGA nicht einmal die Originaldatensätze aus den vorausgegangenen Versuchsreihen der Pharmaunternehmen vor. Nur zwei der befragten Institutionen, die FDA und die japanische Pharmaceutics and Medical Devices Agency PMDA, erhalten die Datensätze aus den Zulassungsstudien, und diese Daten werden entgegen der sonst üblichen good scientific practice grundsätzlich nicht veröffentlicht [3, 16, 17, 18, 19, 20]. Dies wird von Wissenschaftlern offen kritisiert [21]. Gerade im Rahmen beschleunigter Zulassungsverfahren kommt es immer wieder zu Problemen. Verzögerungen bedeuten finanzielle Verluste für die Pharmakonzerne, weswegen teilweise übereilt entschieden wird, zudem sind die Hürden in beschleunigten Zulassungsverfahren geringer [3, 30]. Dies hat dazu geführt, dass der Pfizer-Impfstoff trotz einer Inzidenz von 12,5 ernstzunehmenden Nebenwirkungen auf 10 000 Impfungen zugelassen wurde [22].
Zudem bestehen laut dem Journalisten Paul Thacker Zweifel an der Integrität des Datensatzes, d. h. der Datensatz ist in sich nicht logisch, was darauf schließen lässt, dass die Daten entweder versehentliche Fehler enthalten oder manipuliert wurden [16]. Außerdem gibt es Hinweise auf Fehler in der Studiendurchführung. Qualitätskontrollen bei Pfizer offenbarten erhebliche Probleme, wie etwa, dass das Personal eventuell wusste, welche Probanden* den Impfstoff erhalten hatten und welche den Placebo, weil entsprechende Hinweise offen herumlagen, oder auch dass Probanden nach der Impfung nicht von medizinischem Personal beobachtet wurden [16]. Zudem wurden symptomatische Patienten vor der Impfung wegen Personalmangel nicht in allen Fällen auf Covid-19 getestet. Vom Qualitätsmanagement beanstandete Probleme wurden von Pfizer über Wochen nicht behoben [16]. Auch die FDA hat nicht auf die Beschwerden des Qualitätsmanagements reagiert [23], und auch bei der FDA selbst gibt es entsprechende Probleme. Nur neun der 153 Pfizer-Zentren, in denen die Studie durchgeführt wurde, wurden überhaupt von der FDA kontrolliert [24]. Dies gilt auch für die anderen Impfstoffe. Nur zehn von 99 Forschungszentren von Moderna [25] wurden kontrolliert und fünf von 73 für Remdesivir [26]. Ebenso gibt es von anderer Stelle Berichte darüber, dass Probandinnen* aus ungeklärten Gründen aus der Studie gestrichen worden seien [45]. Auch die erforderlichen Schutzmaßnahmen für Studien an Menschen wurden in der Pfizer-Studie nicht eingehalten [27], und unabhängig von der Pfizer-Studie prüft die FDA nur 1 % aller Einrichtungen, an denen Zulassungsstudien stattfinden, Tendenz abnehmend [28, 29]. Zwischen März und Juli 20 wurden die Besuche komplett eingestellt [23].
Ähnliche Probleme hatte es laut Wissenschaftlern und Journalisten bei den Inspektionen der FDA auch schon vor Corona gegeben [31 zitiert nach 23, 32, 33, 34]. Weiterhin hat die massiv von der Pharmaindustrie finanzierte FDA keinen Überblick über Einrichtungen in den USA und weltweit, an denen medizinische Versuche an Menschen durchgeführt werden [34, 3]. Die FDA habe zudem vermehrt Probleme gehabt, Personal zu rekrutieren und zu halten, die Fluktuation im Personalbereich sei sehr hoch, die Löhne niedriger als in der Privatwirtschaft. Zwei Mitarbeitende hätten während der Pandemie Suizid begangen. Die FDA hat nur 89 Inspektoren für die Hunderttausenden Einrichtungen, in denen Medikamente weltweit für US-Firmen getestet werden, obwohl sie 18 000 Mitarbeitende beschäftigt [35, 36].
Wenn die FDA Probleme wie fehlende Zustimmung der Teilnehmenden, Daten- oder Meldefehler in einem Zulassungsverfahren feststellt, informiert sie zudem nicht zwingend die Fachöffentlichkeit. Veröffentlichte Informationen werden außerdem vor der Veröffentlichung teilweise redaktionell verändert [37, 38]. Im Falle des Blutgerinnungshemmers Rivaroxaban hatte die FDA eine Zulassungsstudie aufgrund schwerer Mängel aus den offiziellen Studien für das Zulassungsverfahren ausgeschlossen. Die Studie wurde später trotzdem in The Lancet veröffentlich und über 1100 mal zitiert. Die Autoren der Studie hatten von der FDA keine Rückmeldung über die Mängel erhalten [39, 40]. Auch im Falle des Antibiotikums Ketek gab es Unregelmäßigkeiten im Zulassungsverfahren. Das Medikament wurde ab 2001 erprobt und in 2004 zugelassen, bis 2007 gab es Hunderte Berichte von unerwünschten Nebenwirkungen, Dutzende Todesfälle und zwei Kongressanhörungen wegen des Medikamentes. Die FDA hatte nur eine der 1800 Forschungseinrichtungen inspiziert und schwere Fehler im Studienverlauf gefunden, so etwa Unregelmäßigkeiten bei der Aufnahme von Probanden in die Studie. Im Rahmen eines Strafverfahrens kam es zu einer Haftstrafe von 57 Monaten für einen der Beteiligten. Aufgrund der laufenden Ermittlungen durften jedoch die Probleme in der Klinik nicht im Rahmen eines FDA-Meetings berichtet werden, das Komitee stimmte also für die Zulassung. Hierbei wurden nur einzelne Meldungen von Nebenwirkungen berücksichtigt, was nicht der gängigen Praxis in Zulassungsverfahren entspricht [41, 42, 43].

Abb. 2: Ansprachen zum Zulassungszeitpunkt (Quelle: https://sashalatypova.substack.com/p/letters-from-the-underworld?utm_source=%2Fsearch%2Flatypova&utm_medium=reader2)
Klinische Zulassungsstudien sind also ein komplexer globaler Markt, und die FDA hat eventuell gar nicht die Ressourcen, um die gesamte Forschung aller US-Konzerne weltweit zu überwachen. Dazu, ob dies auch in einigen Ländern in Europa oder für die Europäische EMA gelten könnte, gibt es ebenfalls Berichte. In Großbritannien etwa hat laut einem Bericht der Wissenschaftsplattform scifiles.org nur der damalige Gesundheitsminister Matt Hancock allein über die Zulassung entschieden [46], aber auch bei der EMA lief das Prüfverfahren nicht problemlos ab [47]. Laut scifiles.org hat Ursula v. d. Leyen für die EU schon vor Erteilung der bedingten Zulassung Impfstoffe bestellt, und die nationale Zulassung, die nach Artikel 5 “RICHTLINIE 2001/83/EG DES EUROPÄISCHEN PARLAMENTS UND DES RATES vom 6. November 2001 zur Schaffung eines Gemeinschaftskodexes für Humanarzneimittel” hätte Anwendung finden können, wurde durch v. d. Leyen explizit verhindert [47, 48, 49]. Absatz 3 des genannten Artikels schließt übrigens zusammengefasst die Herstellerhaftung im Notfall aus, ungeachtet dessen, ob eine gemeinschaftliche oder nationale Genehmigung erteilt wurde, und die Nationalstaaten können die gemeinschaftliche Zulassung später nicht einzeln widerrufen [47]. Laut der Pharmaexpertin Latypova gab es unter den Wissenschaftlern der EMA Sicherheitsbedenken, diese wurden aber von den Kommissaren ignoriert [48, 49]. Eine interne Email zeigt gemäß Latypova, dass gezielt daran gearbeitet wurde, die nationalen Gesundheitsminister zu bewegen, nicht den nationalen Weg zu gehen, so dass die Zulassung schnell gehen könne [48]. Zudem waren die Reviewer der EMA unter massivem politischem Druck, den Impfstoff von Pfizer zuzulassen. FDA, MHRA und EMA hatten den Zeitpunkt der Zulassung bereits vor Durchsicht der Daten untereinander koordiniert (s. Abb. 2) [48, 49]. In Latypovas Dokumenten ist die Rede davon, dass Pfizer sogar falsche Daten übermittelt hätte, um die Zulassungsbehörden zu täuschen [48, 49]. Noch Ende November hatten die Prüfer massive Bedenken bezüglich der Zulassung, wie eine von Latypova veröffentlichte interne Email zeigt (Abb. 3) [10].

Abb. 3: Bedenken im Zulassungsverfahren noch kurz vor der Zulassung (Quelle: https://sashalatypova.substack.com/p/letters-from-the-underworld?utm_source=%2Fsearch%2Flatypova&utm_medium=reader2)
Auch eine Gruppe deutscher Juristen äußert sich kritisch zur Zulassung der neuartigen Corona-Impfstoffe [30]. So handele es sich aus juristischer Sicht nicht um eine Impfung, da die Präparate nicht wie in der EU-Richtlinie zu Impfungen definiert Antigene sind, sondern einen Bauplan für Teile des Virus enthalten [30]. Genbasierte Medikamente würden laut den Juristen höheren Standards unterliegen als Impfstoffe. Die EU-Kommission hätte allerdings in 2009 nach einer Stellungnahme der Pharmaindustrie Impfstoffe gegen Infektionskrankheiten aus der Gruppe der besonders regulierten Gentherapeutika herausgelöst. Nach dem ursprünglichen Richtlinienentwurf wären die neuartigen Corona-Impfstoffe noch nach den Regelungen für Gentherapeutika gehandhabt worden [30]. Nachdem die Pharmaunternehmen sich dahingehend geäußert hätten, dass die Anwendung der Richtlinie auf diese Impfstoffe die Produktion verteuern würde, sei die Regelung geändert worden, so dass die Impfstoffe nicht mehr darunter fielen [30]. Im nächsten Schritt folgte dann die Umwandlung der Notfallzulassung in eine reguläre Zulassung [30]. Hierzu wären eigentlich mehrjährige Placebo-kontrollierte Doppelblindstudien erforderlich, diese dauern aber und sind teuer. Nach der Umwandlung in eine reguläre Zulassung hätten die Pharmaunternehmen nunmehr keinen Anreiz mehr, derart teure Studien durchzuführen [30]. Die EU-Kommission hat damit laut den Juristen gegen Art. 14-a Abs. 8 der Verordnung Nr. 726/2004/EG und Art. 7 der Kommissionsverordnung Nr. 507/2006/EG verstoßen, welche genau solche Studien vorschreibt [30]. Ohne diese Art von Studien dürfe die reguläre Arzneimittelzulassung laut Art. 12 Abs. 1 der Verordnung 726/2004/EG nicht erteilt werden [30]. Zudem hatte Pfizer/Biontech die Kontrollgruppe aus der Studie gelöst, so dass auf diesem Wege keine Langzeitdaten zur Impfstoffsicherheit mehr erhoben werden können, entsprechende Studien seien also kaum noch möglich [30].
Es muss daher festgestellt werden: jede Prüfung ist nur so gut wie der, der sie bezahlt…
7 Antworten
Na das erklärt auch, warum die Zulassung von Sputnik V im Nichts versickerte…
Umso mehr diese Firmen pro Jahr mit patentierbaren Substanzen verdienen, umso mehr können sie weltweit Manipulationen jeglicher Art durchführen. Selbst Milliarden Strafen planen sie schon mit ein. Es ist unerträglich, was sich da mit den Jahren entwickelt hat und abspielt. Ganz übelst ist es dann zu beobachten, wie sie überall in den Medien versuchen, natürliche, lebensnotwendige, nichtpatentierbare Substanzen verunglimpfen. Nur um das Geschäftsmodell mit der Krankheit, weiter aufrechtzuerhalten. Mit allen erdenklichen Tricks manipulieren sie seit Jahren Gesetze und setzen sie wirkungsvoll weltweit durch. Wenn wir weiter diese Strategie verfolgen, dann steigen die Kosten ins Uferlose, denn dieses Krankheits-Unwesen ist jetzt schon unbezahlbar geworden. Man beachte nur die jährlichen steigenden Ausgaben in dem Bereich.
Was für eine Sauerei! Und so viele Menschen sind an diesen Prozessen beteiligt… Was für eine Schmach sie sich da wohl aufgeladen haben? Oh je…
Toller Artikel. Danke, dass Sie Licht ins Dunkel bringen!
Danke
Ich würde gerne folgende persönliche Überlegungen in einen ergebnisoffenen Diskursraum stellen:
Ich beobachte mit großer Freude,
dass die Menschen nach wie vor, mal mehr und mal weniger, eigene Überlegungen zu bestimmten Lebensbereichen anstellen und bei Ihren Überlegungen, neben persönlichen Prägungen, persönlichen Beobachtungen, persönlichen Erlebnissen und persönlichen Recherchen, das jeweilige auserkorene Erkenntnisobjekt auf seinen Ursprung zurückzuverfolgen versuchen.
Wissend, dass sich jeder Mensch von seinen Mitmenschen unterscheidet, sprich: unterschiedliche Prägungen, Erlebnisse, Sozialisation, Wissen, Fähig- und Fertigkeiten, etc. im Laufe seines eigenen Seins erfahren hat, gilt es bei Analysen zu jedem möglichen Erkenntnisobjekt zu berücksichtigen, dass es Erkenntnisobjekte gibt, welche auf eigenständig überprüfbaren Fakten und ebenso andere auf reinen Glaubenssätzen beruhen.
Jeder Mensch kann eigenständig die These der Schwerkraft überprüfen und wird unabhängig von Zeit und Ort immer dieselbe Erkenntnis erlangen, ob er an die Schwerkraft glaubt oder nicht.
Medikamente, wie in dieser Recherche ansatzweise beleuchtet, sind nur bedingt bis gar nicht von jedem Menschen auf Erkenntnisgewinn zu überprüfen. Damit ist die Entwicklung, Empfehlung, der Verkauf, die Verabreichung und der Konsum von Medikamenten mit großen und nicht selten sogar lebensbedrohlichen Risiken versehen.
Jeder Mensch, der verstanden hat, dass sein eigener Körper sein einziges Grundkapital ist, mit dessen Zuhilfenahme (Mittel) er bei Funktionalität des Körpers überhaupt erst handlungs- und somit lebensfähig (Gütertausch) wird und ist, hat auch verstanden, dass die Gesunderhaltung des eigenen Körpers essentiell für sein gesamtes Leben ist und bis zum letzten Tag sein wird.
Jeder Mensch, der die Funktionsweise der Wirtschaft verstanden hat weiß, dass jeder Mensch immer gewinnorientiert handelt, sprich: mit seiner Handlung seinen aktuellen Zustand zu verbessern oder zumindest eine Verschlechterung zu vermeiden versucht. Selbst Altruisten und Masochisten streben einen Gewinn mit ihren Handlungen an. Die Logik des menschlichen Handelns ist nicht nur unglaublich erkenntnisreich, sondern insbesondere völlig wertfrei.
Wissend, dass jeder Mensch an seinen eigenen Körper gebunden ist und ausschließlich mit seinem eigenen Körper als Mittel selbständig und damit eigenverantwortlich handeln kann, sollte unumstritten klar sein, dass jeder Mensch qua Geburt der Exklusiveigentümer seines eigenen Körpers ist und immer bleibt, auch dann, wenn andere Menschen das nicht respektieren und aggressiv über eine feindliche Handlung mit ihrem Körper auf den Körper eines Mitmenschen zugreifen.
Diese einfache und logische Schlussfolgerung führt jeden selbständig denkenden Menschen automatisch zu der Erkenntnis, dass kein Mensch gegen den Willen eines anderen Menschen über den Körper des anderen Menschen bestimmen und verfügen können darf. Das würde ansonsten nichts anderes als die Versklavung eines Menschen durch einen anderen Menschen bedeuten, was eigentlich von den meisten Menschen im 21. Jh. abgelehnt und als unmenschlich, unanständig und ein Verbrechen angesehen und daher abgelehnt werden sollte. Dem ist jedoch in der Realität mit Nichten der Fall.
Wäre dem so,
dann wäre es KEINEM Menschen erlaubt:
– Mitmenschen zu berauben
– dem Körper von Mitmenschen Schaden zuzufügen
– Mitmenschen zu entführen
– Mitmenschen die Nutzung von Sprache zu verbieten
– etc.
sondern JEDER Mensch hätte das Recht, sich gegen jedweden aggressiven und feindlichen Angriff auf sein Privateigentum zu verteidigen, ggfs. mit denselben Mitteln, die der Aggressor für seinen feindlichen Angriff verwendet.
Was passiert, wenn manche Menschen an Mitmenschen ihrer Wahl Verbrechen verüben dürfen und die Opfer sich weder gegen den Aggressor erwehren noch zivilisiert Wiedergutmachung, Vergeltung, Schadensersatz oder auch Vergebung von dem Täter erwirken dürfen, kann jeder Mensch eigenständig überprüfen und tagtäglich beobachten.
In diesen wenigen, einfachen und für jeden Menschen eigenständig immer, überall und zu jeder Zeit überprüfbaren Fakten finden Sie nicht nur die Ursachen für Leid, Elend und Tod von vielen Menschen und eine regelrechte Anreizstruktur von Verbrechen für immer mehr empathie-, verantwortungs- und herzlose Mitmenschen, sondern auch die Lösung.
…die swissmedic wurde verklagt…
https://www.youtube.com/watch?v=AJCGCe8bkis
Das wird man gegenüber der EMA wohl eher nicht schaffen?…Ich glaube gern, daß dort ein erheblicher politischer Druck geherrscht hat. Ein einziges französisches Institut hat veröffentlicht: “Die Imofstoffe sind nicht wirksam und nicht sicher”…es wurde nicht gehört..
Ergänzend dazu die von RA Philipp Kruse dazu eingerichtete Netzseite: https://coronaanzeige.ch